

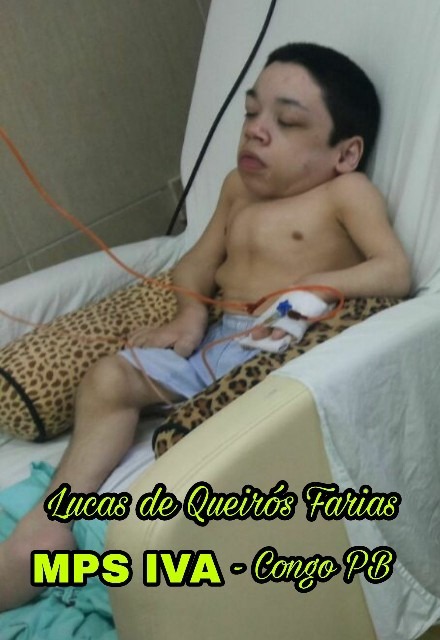
Lucas de Queirós Farias, da cidade do Congo na Paraíba faleceu nesta quinta-feira (8) , o jovem de 26 anos era paciente de MUCOPOLISSACARIDOSE, onde conseguiu uma Liminar assinada pela justiça Brasileira que lhe dava direito a um tratamento que o mantinha vivo .
Lucas Iniciou terapia de reposição enzimática em 2014; revoltantemente o mesmo perdeu a liminar, Sua última infusão foi em 7 de março de 2017, de lá para cá, todas as suas funções vitais decaíram e ele ficou praticamente tetraplégico, cego e surdo. Lucas estava há quase 2 anos sem medicação, quando seu tratamento foi interrompido por um juiz.
Notícias Relacionadas

Greve
Alunos do IFPB fazem protesto e pedem volta às aulas
Professores e servidores técnico-administrativos estão de greve desde o dia 3 de abril.

Transplantes
Central de Transplantes registra duas doações de multiórgãos no mesmo dia e 11 vidas serão transformadas
Em 2024, já foram registrados a renovação de 89 vidas por meio da doação de órgãos.
Há 1 ano, quase nesta mesma época, seu irmão MATHEUS faleceu, também sem medicação, porém foi em virtude do descumprimento de ordem judicial pelo então Ministro da Saúde Ricardo Barros. Matheus de Queirós Farias Paraibano, 23 anos, e também paciente portador de mucopolissacaridose
O que é são as Mucopolissacaridoses
As Mucopolissacaridoses (MPS) são doenças metabólicas causadas por erro inato do metabolismo. Estes erros levam à formação inadequada de enzimas, substâncias fundamentais para diversos processos químicos em nosso organismo. As enzimas que não funcionam adequadamente nas são encontradas nos lisossomos (estrutura que funciona como uma usina de reciclagem nas células).
No paciente com MPS, os lisossomos não conseguem cumprir corretamente sua função, que é digerir grandes moléculas para serem utilizadas ou reutilizadas. No caso desta doença, tais moléculas mal digeridas são os Glicosaminoglicanos (GAGs), responsáveis pela lubrificação e união entre os tecidos. Daí uma consistência mucoide, viscosa.
Tais partículas (GAGs) se acumulam no lisossomo, fazendo as células crescerem. Outra parte é eliminada pela urina. Com isso ocorre o acumulo em órgãos como a pele, o fígado e o baço. Portanto, as MPS são também consideradas doenças de depósito lisossômico.
Há diversos tipos de Mucopolissacaridoses, que diferem de acordo com a enzima que está em falta do organismo do paciente. Foram identificados ao topo sete tipos de MPS (no portal Muitos Somos Raros, você poderá conhecer melhor todos os tipos).
Causa
As MPS têm origem genética e hereditária. Como a maioria das doenças metabólicas, elas têm herança de caráter autossômico recessivo. Ou seja, os pais são sadios, mas carregam um gene que determina a doença, e que só se manifesta em dose dupla. Assim, os pais portadores têm, a cada gestação, 25% de chance de ter um filho que desenvolva a MPS (e 75% de ter um filho sem a doença, sendo que 50% podem também carregar o gene, mas sem desenvolver a doença).
A exceção é a MPS tipo II (Síndrome de Hunter), com herança ligada ao cromossomo X. Neste caso, a mãe portadora (sadia mas que carrega o gene tem 50% de chance de ter um filho homem com a doença, e 50% de ter uma filha que carregue o gene da doença para seus descendentes).
Fonte: Polêmica Paraíba
Créditos: Polêmica Paraíba





